SaaS
NEO-ARS® is a comprehensive service that identifies tumor neoantigens and covers the whole process from NGS data analysis to 3D structure-based prediction of peptide-MHC (pMHC) binding using multiple AI algorithms.
Top 20 immunogenic neoantigen candidates are ranked as promising targets for cancer immunotherapy.
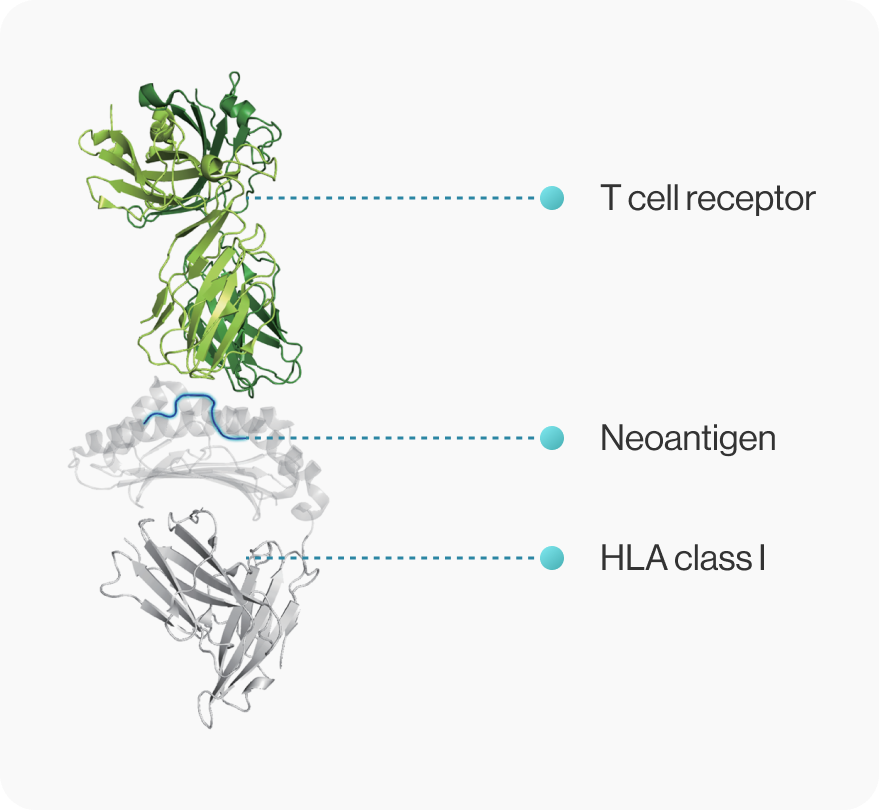
Cancer vaccine developers want to obtain 3D structural information about how genetic mutations occurring in self-peptides yield immunogenic peptides that stably bind to MHC molecules.
NEOscan™ predicts both pMHC binding energy and T cell reactivity based on known crystal structures
Many clinicians and biotech companies interested in personalized cancer immunotherapy hope to discover neoantigen candidates as promising targets for cancer vaccines and T cell therapies.
NEO-ARS® i) determines the patient's HLA type, ii) prioritizes genetic mutations considering several parameters (e.g., sequencing depth, VAF(%), RNA expression level), and iii) identifies neoantigen candidates. It covers everything from genome profiling to in silico analysis of the 3D binding structure of pMHC complex.
A. Workflow

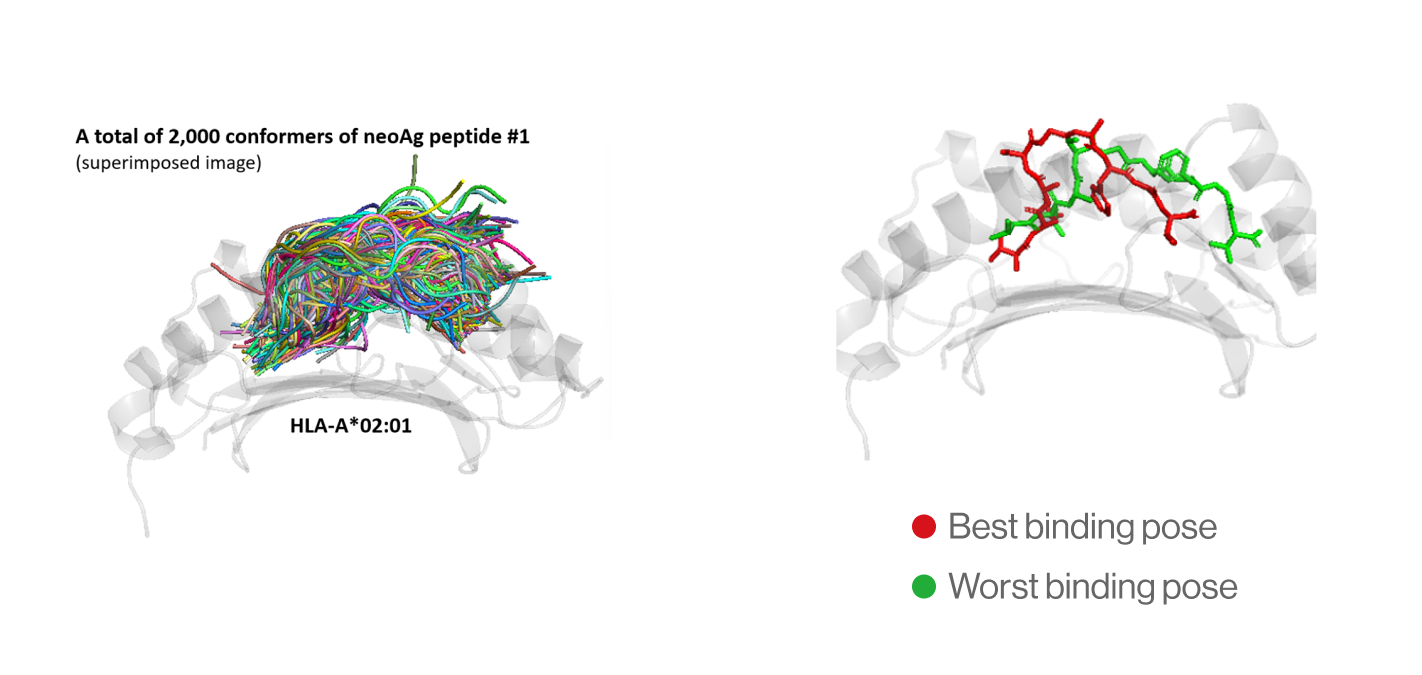
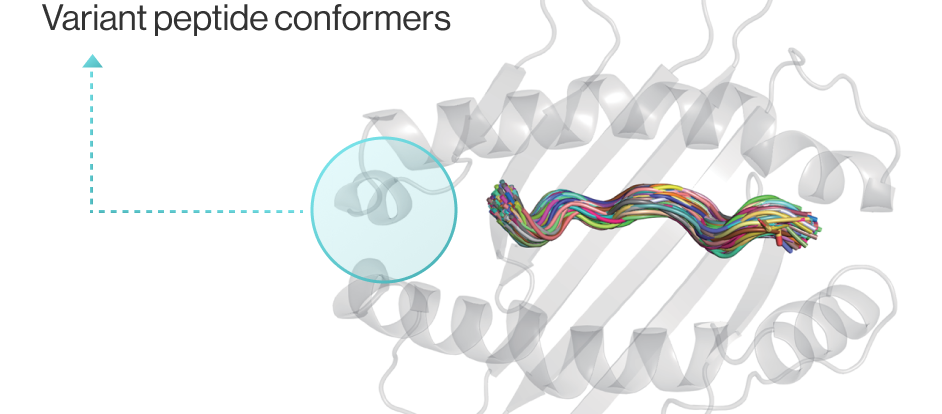
B. Selection of best binding pose
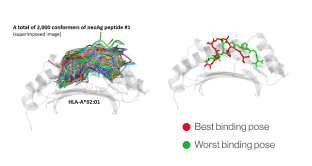
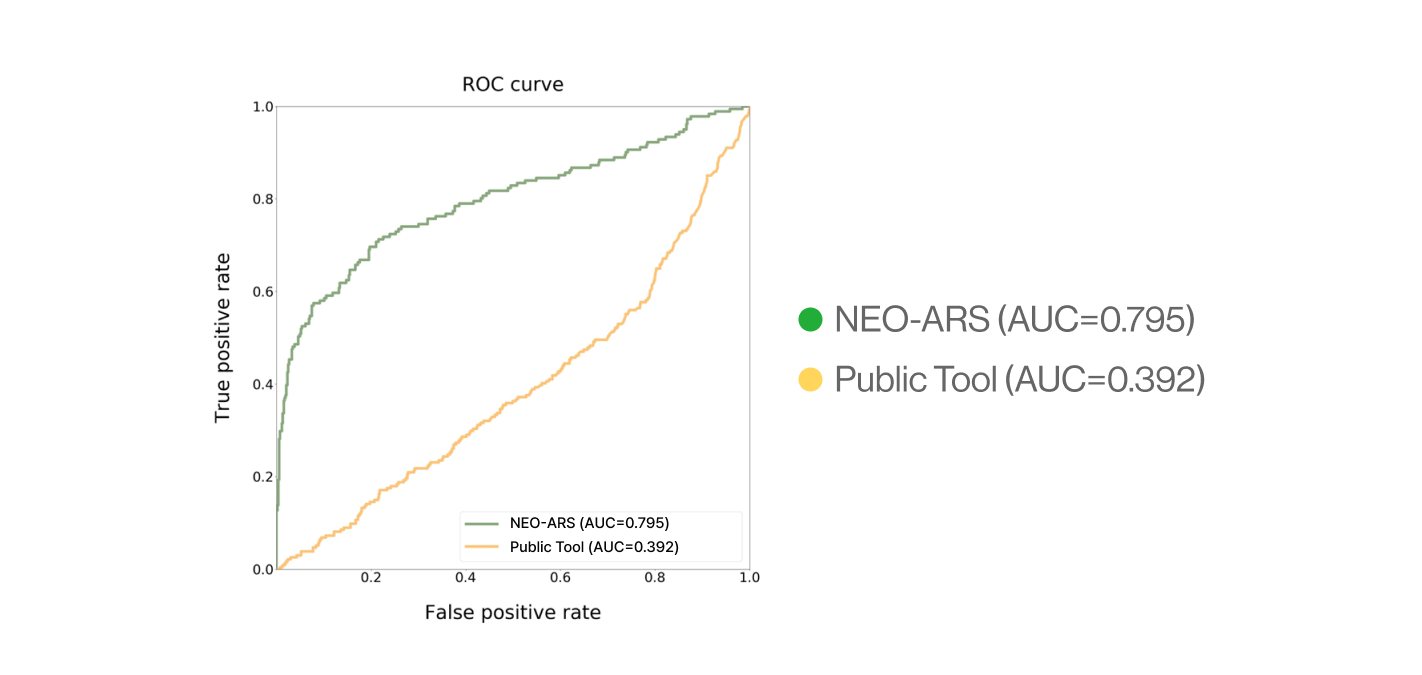
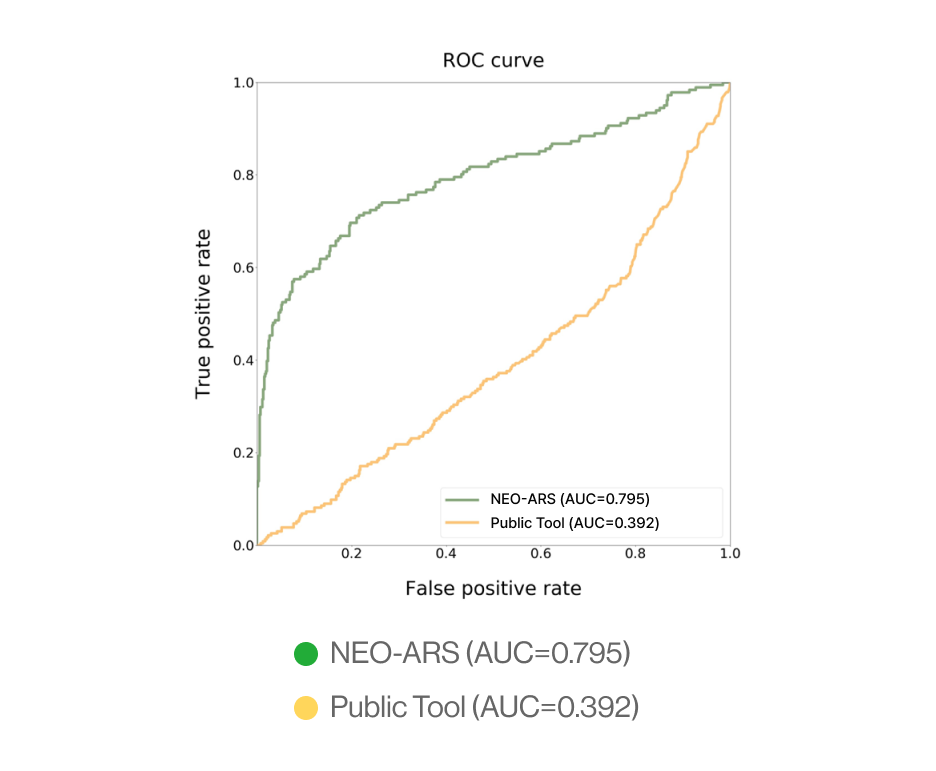
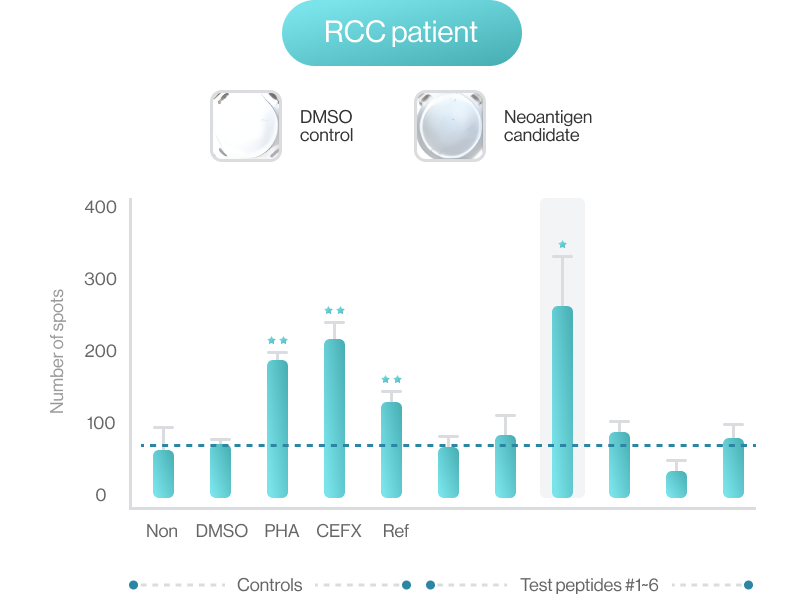
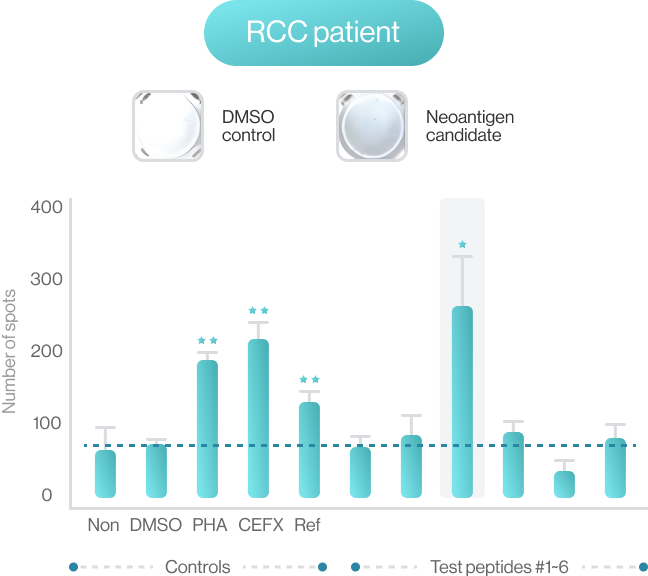
C. External validation
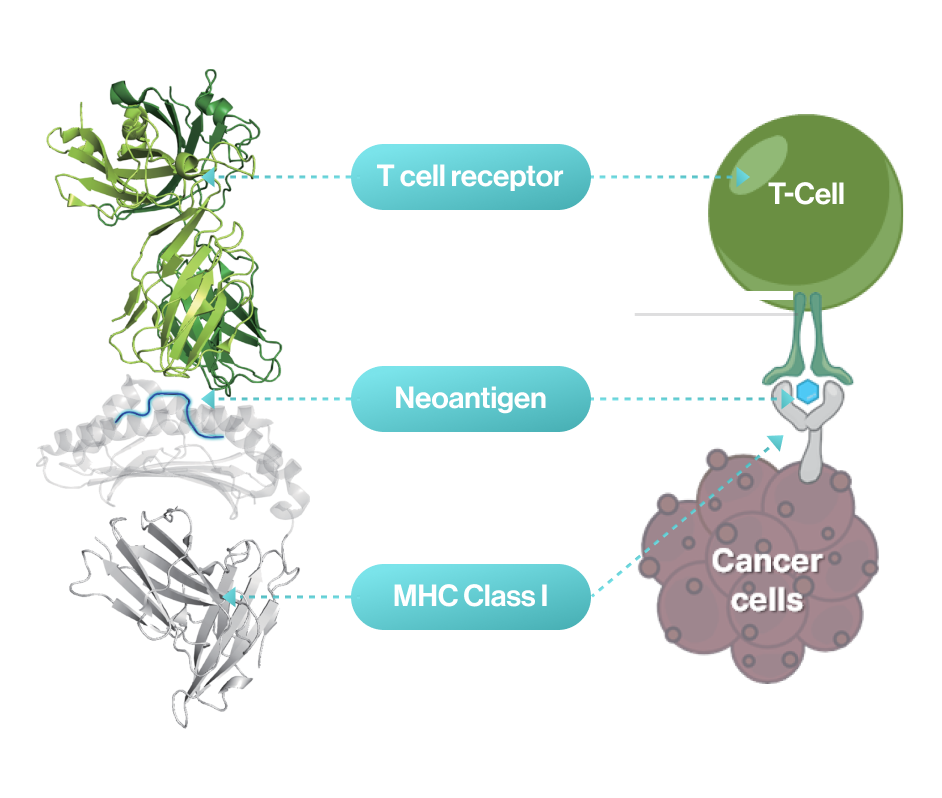
About NEO-ARS®
NEO-ARS® predicts peptide-MHC binding pattern of immunogenic neoantigen based on 3D structure.

Conventional Tool
NEO-ARS®
Conventional Tool
NEO-ARS®
Conventional Tool
-
NEO-ARS®
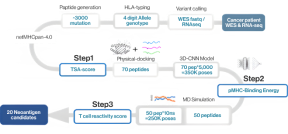
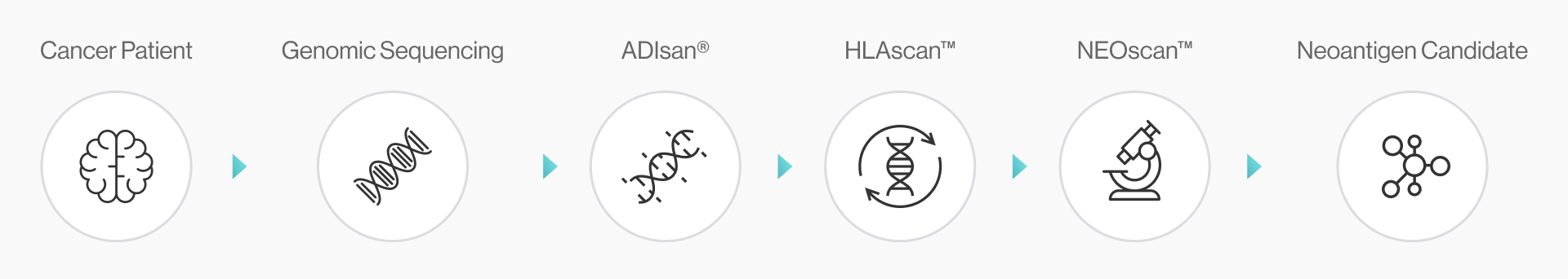
NEO-ARS® neoantigen prediction process is applied in two steps: the first one is NGS pipeline; the latter is NEO pipeline.
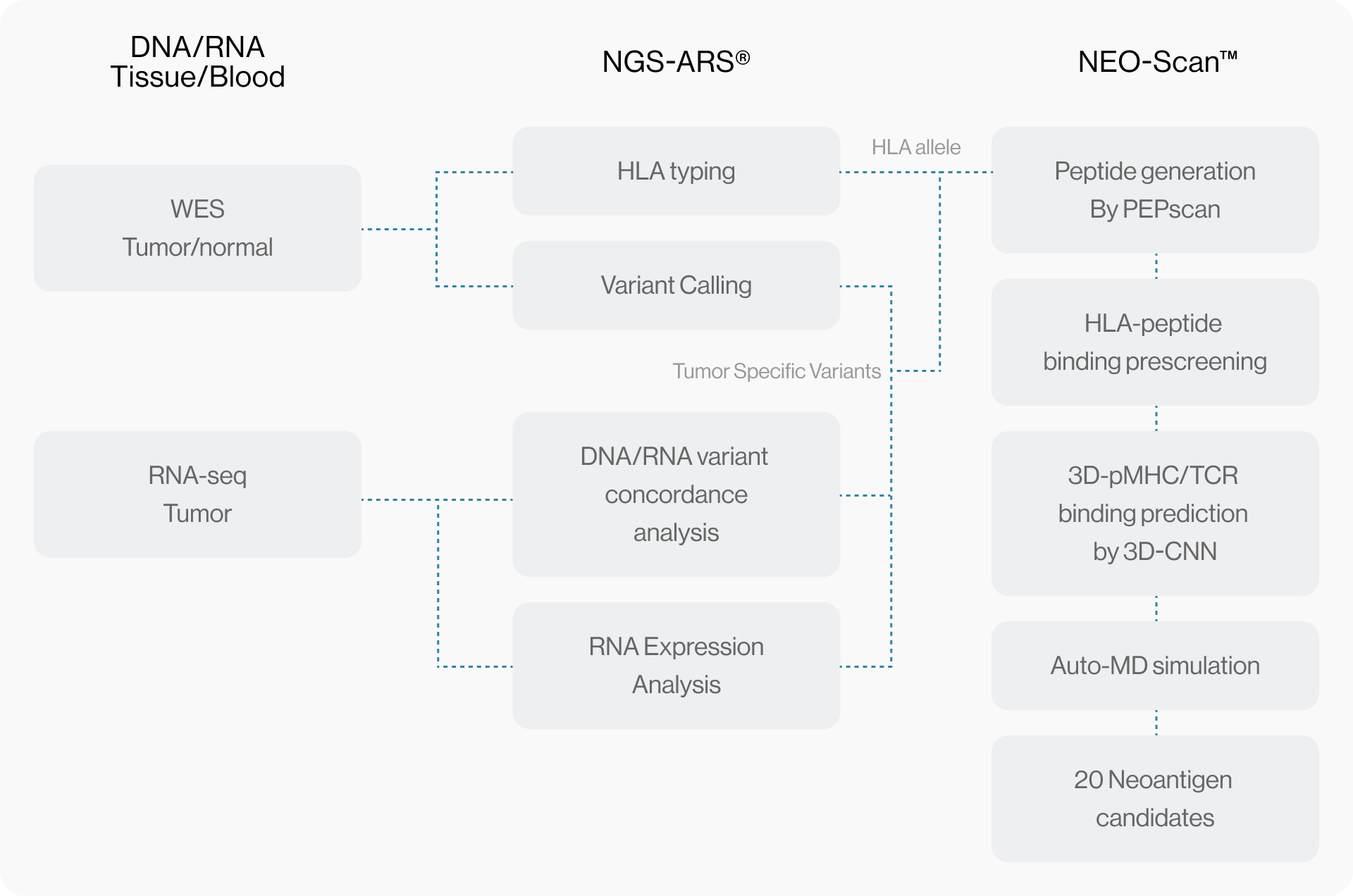
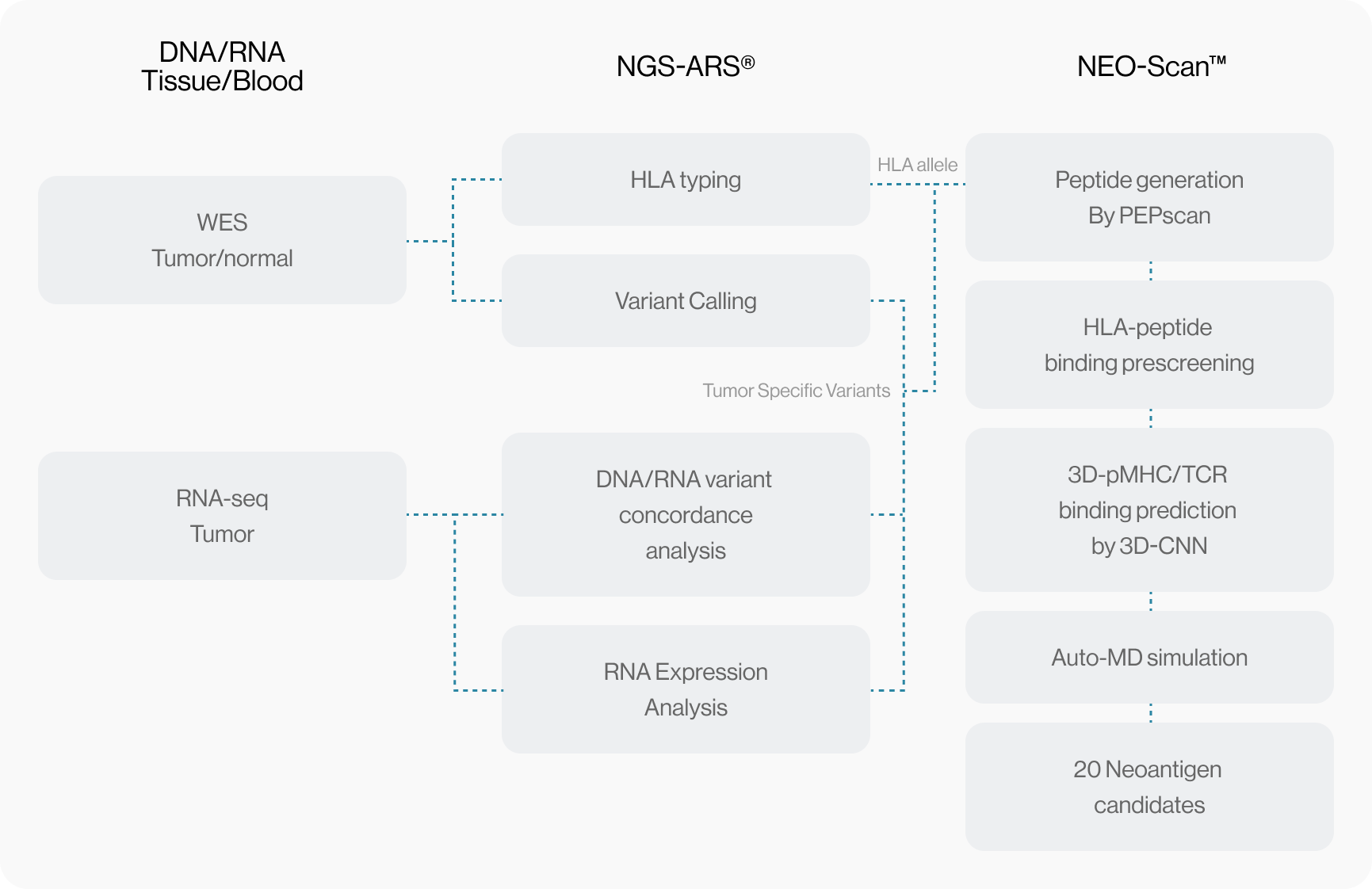
NGS pipeline, NEO-ARS® is an optimized algorithm to analyze tumor specific somatic variants and HLA typing based on bioinformatics of genomic data integrated between whole exome sequencing (WES) and transcriptome sequencing.
NEO pipeline, AI-based prediction methods with 3D-CNN model for Peptide-MHC binding, key-residue model for TCR-peptide binding and MD simulation can predict physicochemical properties and dynamic T cell responses.
Our maximized neoantigen prediction platform provides a comprehensive solution between tumor mutations based therapeutic approaches.
NEO-ARS® predicts peptide and MHC binding based on 3D-CNN AI model and performs fine-tuning through MD simulations to provide more accurate and rigorous calculations. The selected candidate peptides are effectively narrowed down to those more likely to induce an immune response, selecting more meaningful candidates for subsequent experimental validation.
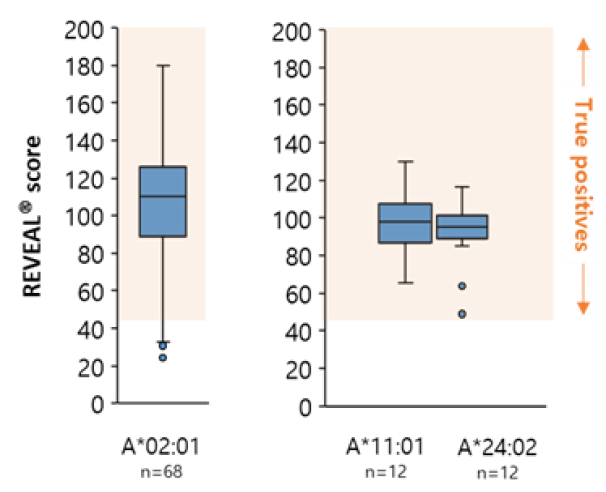
AI-model performance evaluationusing in vitro peptide-HLA binding assay (previous study 2022’)
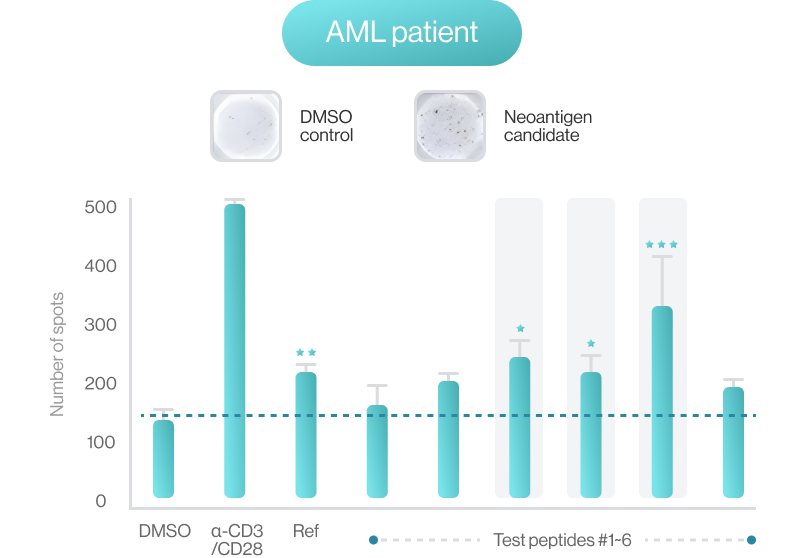

NEO-ARS® predicts peptide and MHC binding based on 3D-CNN AI model and performs fine-tuning through MD simulations to provide more accurate and rigorous calculations. The selected candidate peptides are effectively narrowed down to those more likely to induce an immune response, selecting more meaningful candidates for subsequent experimental validation.


Definition
Definition
Definition
This program is in-silico prediction tool, so we recommend proceeding with next-stage experiments such as immunogenicity or binding affinity tests with these neoantigen candidates
It covers the whole process from NGS analysis to neoantigen prediction.. NEO-ARS® neoantigen prediction process is applied in two steps: the first oneis NGS pipeline; the latter is NEO pipeline
Process
Input
WES pairedtumor/
normal tissue,
RNA-seq with tumor
tissue,
Fastq or BAM
Runtime
~5 days per 1 run
Output
9-mer’s 20 neo-
peptide list
Support
Computer capacity :
I Unit of 1 CPU/1 GPU
Expo-ro 1, Expo Tower #1903,
Yuseong-gu, Daejeon,
Republic of Korea
Headquarter
Samunan-ro 92, Gwanghwamun Officia Building #1708,
Jongno-gu, Seoul, Republic of Korea
Business Center
425 Fifth Avenue Suite 505 New York,
NY, USA #10016
Syntekabio USA Inc.
![]()
© 2024 Syntekabio, Inc. All rights reserved.


