SaaS
We introduce an advanced approach (auto-Best pose) to predict the most likely binding poses of ligands within target proteins, which analyzes binding modes while accounting the integration of structural information, consideration of ligand and protein flexibility, and reliable scoring and evaluation metrics. A fully automated CLOUD SaaS process that can improve efficiency, consistency, scalability, accessibility, and integration with AI algorithms are significant contribution to modern drug discovery efforts. It plays a valuable role for researchers and pharmaceutical companies in identifying promising drug candidates effectively and swiftly.
Researchers found a hit and its binding protein through experiments including cell-based essays, and many other methods, but they cannot proceed its lead optimization because they do not know how it binds.
Auto-BP service is appropriate. For Auto-BP, it is recommended to select low, intermediate or advanced levels depending on the importance of the customer’s lead compound.
A molecular researchers found it certain that it plays a role in cell death, but it is unknown which protein it binds to. What service is appropriate in this case?
MOA (mode of action) service is appropriate. In this case, Auto-BP is set lightly, and instead of performing Auto-BP one molecule against one target 2,000 panel targets are screened simultaneously and a list of suitable candidates is givenas an output. (https://cloud.syntekabio.com)
The AI algorithms of our precision in-house scoring system, are key components underline our screening protocol enabling accurate prediction of binding affinity and orientation. The platform has fully automated all processes from the maximum collection of molecular docking to 3D-CNN and MD simulation for enhanced accuracy and performance in scoring and ranking.

APocket model (~100 templates)
B3D-CNN Model

CMD-simulation, Strain & Binding
Free Energy

DAuto-BP Accuracy
About Auto-Bestpose(BP) Solution

Auto-Bestpose
Application
Key models and methods including
Deliverables
Advantages of our auto-BPservices:
Our high-performance bio-supercomputing processes, synergistically integrated with our cutting-edge SaaS software, proficiently manage extensive calculations with heightened precision. Our software facilitates the execution of high-quality customized projects, encompassing advanced computing simulations and professional drug design, spanning from preliminary assessments to result validation.
Bestpose SCR
Auto-BP
Docking-module
STB Virtual library
Zinc SCR (1B)
DMC-Zinc-Hit
SCR-Docking-module
1B Zinc library
LFS HIT-SCR (50M)
DMC-QSL-Hit
SCR-Docking-module
50M Synplelibrary
FDA drug SCR (10K)
DMC-DR-Hit
SCR-Docking-module
10K FDA drug
Hit-MOA-Kinases
600-Kinase-profile
SCR-Docking-module
Kinase targets
Hit-MOA-Membrane
400-Mem-profile
SCR-Docking-module
Membrane targets
Hit-MOA-Targets
2000-Target-profile
SCR-Docking-module
All known targets

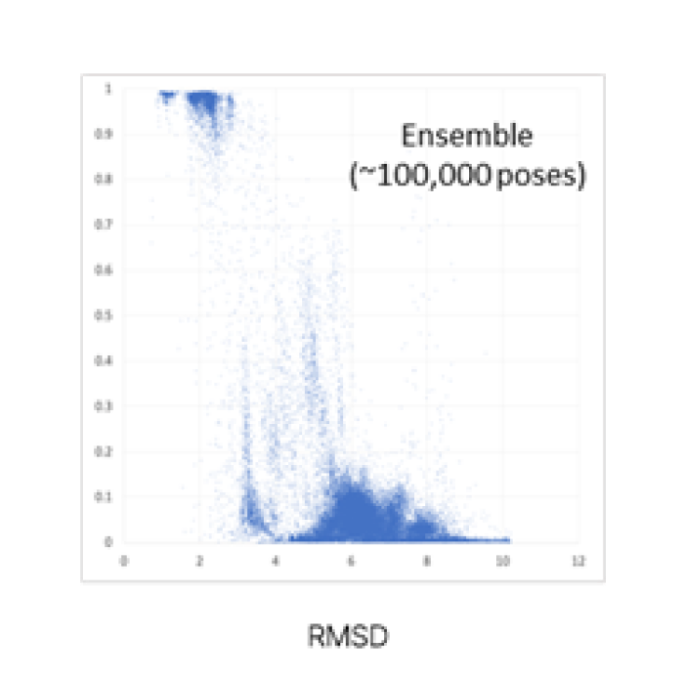
Auto-BP Accuracy
J. Chem. Theory Comput. 2021,17,4 2630-2639

This program is in-silico prediction result, so werecommend proceeding with next-stage experiments such as in-vitro validationtests with these Best-pose candidates
Identify the best-pose to predict the Interaction between target and ligand. Main processes run automatically from screening to 3D-CNN & MD simulation. It generates more than 40,000 conformers as potential poses, select the best-pose
Process
Input
Target +ligand
Runtime
2~3 weeks per 1 run
Output
10 Best-pose with
B.E. value
Support
Computer capacity :
I Unit of 100 CPU
/100 GPU
Expo-ro 1, Expo Tower #1903,
Yuseong-gu, Daejeon,
Republic of Korea
Headquarter
Samunan-ro 92, Gwanghwamun Officia Building #1708,
Jongno-gu, Seoul, Republic of Korea
Business Center
425 Fifth Avenue Suite 505 New York,
NY, USA #10016
Syntekabio USA Inc.
![]()
© 2024 Syntekabio, Inc. All rights reserved.


